病因学
PNH的发病原因有两种:造血干细胞内出现体细胞突变。[3] 有骨髓发育不良的背景。[4]体细胞突变多发生于磷脂酰肌醇多聚糖锚着点的基因上,它可以编码合成磷脂酰肌醇多聚糖锚着点的酶。由于异常细胞中不能合成锚蛋白,可以出现细胞表面锚蛋白全部缺失的细胞(III型PNH细胞),或部分缺失(II型PNH细胞),特别补体是防御蛋白CD55和CD59的缺失。[5]除了基因在复制过程中转录差错未能修复导致的自然发生的突变外,尚没有其他原因能导致这种体细胞的突变。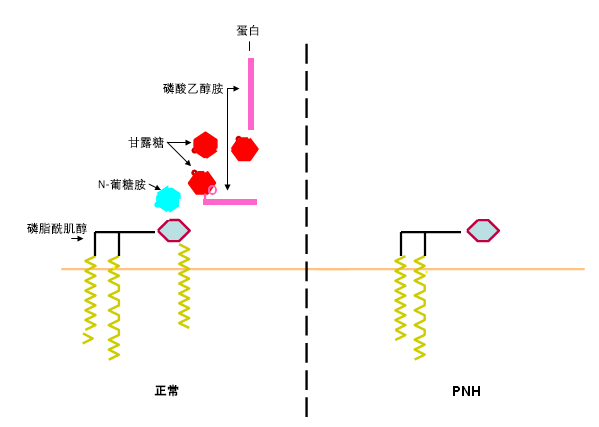 [Figure caption and citation for the preceding image starts]: PNH的糖基磷脂酰肌醇的锚磷脂来自作者Wendell F. Rosse博士 [Citation ends].
[Figure caption and citation for the preceding image starts]: PNH的糖基磷脂酰肌醇的锚磷脂来自作者Wendell F. Rosse博士 [Citation ends].
如果要像再生障碍性贫血那样出现临床症状和抑制造血,有缺陷的克隆必须足够大。[6]在大部分病例中,这可能是源于自身免疫病(即,T 细胞介导的对骨髓的攻击),但也发现过药物和化学制剂诱导的骨髓发育不全的病例。特发性骨髓再生不良的病因目前尚不能确定。
病理生理学
经典的PNH的病生理学改变是,缺乏CD55、特别是CD59的红细胞的补体被激活。[1]因此,微小的疾病一直在活动(引起持续而少量的溶血)。由于溶菌素复合体被定位于异常红细胞上,使得夜间疾病活动加重(引起夜间发作)。对于红细胞,由于细胞膜的破损及细胞的溶解发生于血管内,导致了血红蛋白释放入血浆,如果量足够大,可以出现血红蛋白尿。补体可被感染、手术或创伤激活,从而出现阵发性的红细胞溶解;也可能由于其他尚不明确的原因导致疾病阵发性发作。红细细胞溶解的数量取决于异常红细胞的数量(克隆程度)、细胞的异常及补体激活的程度。
血红蛋白血症易于结合组织中的一氧化氮,这会引起平滑肌收缩,表现为食管痉挛及腹痛等症状。[7]一氧化氮的缺乏可以引起血管痉挛,导致勃起功能障碍,肺动脉高压及肾功能不全。大部分患者出现异常的“乏力”,在排血红蛋白尿期间可加重。血红蛋白尿可以引起急性肾功能不全,短期大量或长期的血红蛋白尿都可以引起肾功能衰竭。[8]
异常血小板表面的补体被激活后导致血小板活化,和其他原因一起可以导致血栓形成,主要是静脉血栓的形成。[1][9]血栓形成的部位大多不寻常,如肝静脉、腹部静脉或大脑静脉窦,同时也可以出现在深静脉以及偶尔出现在动脉中。
分类
推荐的分类[2]
经典溶血性PNH:克隆规模大到足以发现导致溶血的临床证据,而没有其他骨髓疾病的证据。
合并其他特定骨髓疾病的PNH,例如再生障碍性贫血-PNH(确诊再生障碍性贫血并找到PNH证据),以及骨髓增生异常综合征-PNH(确诊骨髓增生异常综合征并找到PNH证据)。
亚临床型PNH:克隆规模小到不足以出现溶血的证据。
使用此内容应接受我们的免责声明。
