大多数类型 PA 的病因尚不明确。家系内发生PA 提示其至少有一些类型的PA具有某种的遗传基础。[26]Gordon RD, Klemm SA, Tunny TJ, et al. Primary aldosteronism: hypertension with a genetic basis. Lancet. 1992;340:159-161.http://www.ncbi.nlm.nih.gov/pubmed/1352575?tool=bestpractice.com一种显性遗传的、糖皮质激素可缓解的 PA变异类型,是 I型家族性醛固酮增多症(FH-I)。FH-I 于 1966 年首次记录,[38]Sutherland DJA, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J. 1966;95:1109-1119.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=4288576http://www.ncbi.nlm.nih.gov/pubmed/4288576?tool=bestpractice.com它由融合合基因引发,其由11-β-羟化酶基因 (CYP11B1) 5' 端的序列和醛固酮合成酶基因 (CYP11B2) 3' 端的序列组成。[39]Lifton RP, Dluhy RG, Powers M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355:262-265.http://www.ncbi.nlm.nih.gov/pubmed/1731223?tool=bestpractice.com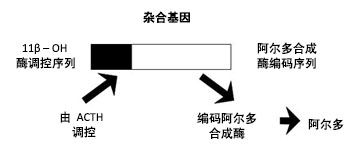 [Figure caption and citation for the preceding image starts]: 在 FH-I 中,CYP11B1/CYP11B2 融合基因导致促肾上腺皮质调节的醛固酮过量产生来自 Michael Stowasser 博士的个人收集;经获准使用 [Citation ends].
[Figure caption and citation for the preceding image starts]: 在 FH-I 中,CYP11B1/CYP11B2 融合基因导致促肾上腺皮质调节的醛固酮过量产生来自 Michael Stowasser 博士的个人收集;经获准使用 [Citation ends].
在 1991 年,[40]Gordon RD, Stowasser M, Tunny TJ, et al. Clinical and pathological diversity of primary aldosteronism including a new familial variety. Clin Exp Pharmacol Physiol. 1991;18:283-286.http://www.ncbi.nlm.nih.gov/pubmed/2065471?tool=bestpractice.com第二种被记录家族性 PA (FH-II),[9]Stowasser M, Gordon RD, Tunny TJ, et al. Familial hyperaldosteronism type II - five families with a new variety of primary aldosteronism. Clin Exp Pharmacol Physiol. 1992;19:319-322.http://www.ncbi.nlm.nih.gov/pubmed/1521363?tool=bestpractice.com其既不是糖皮质激素可缓解性也非融合合基因无关。醛固酮生成腺瘤以及双侧肾上腺增生 (BAH)通常可出现在同一个家族内。[3]Gordon RD, Stowasser M, Klemm SA, et al. Primary aldosteronism and other forms of mineralocorticoid hypertension. In: Swales JD, ed. Textbook of hypertension. London: Blackwell Scientific; 1994:865-892.[10]Gordon RD, Stowasser M. Familial forms broaden horizons in primary aldosteronism. Trends Endocrinol Metab. 1998;9:220-227.[18]Stowasser M, Gordon RD. Familial hyperaldosteronism. J Steroid Biochem Mol Biol. 2001;78:215-229.http://www.ncbi.nlm.nih.gov/pubmed/11595502?tool=bestpractice.com已报告 FH-II 的发病率至少比 FH-I 高 5 倍。[19]Stowasser M, Gordon RD. Primary aldosteronism: from genesis to genetics. Trends Endocrinol Metab. 2003;14:310-317.http://www.ncbi.nlm.nih.gov/pubmed/12946873?tool=bestpractice.com从临床、生化和形态学上,FH-II 与散发性 PA难以区分。[10]Gordon RD, Stowasser M. Familial forms broaden horizons in primary aldosteronism. Trends Endocrinol Metab. 1998;9:220-227.[18]Stowasser M, Gordon RD. Familial hyperaldosteronism. J Steroid Biochem Mol Biol. 2001;78:215-229.http://www.ncbi.nlm.nih.gov/pubmed/11595502?tool=bestpractice.com[19]Stowasser M, Gordon RD. Primary aldosteronism: from genesis to genetics. Trends Endocrinol Metab. 2003;14:310-317.http://www.ncbi.nlm.nih.gov/pubmed/12946873?tool=bestpractice.com,因此,导致 FH-II 的基因突变可能是导致其他无此病家族史的PA患者的主要原因。[5]Gordon RD, Stowasser M, Klemm SA, et al. Primary aldosteronism: some genetic, morphological, and biochemical aspects of subtypes. Steroids. 1995;60:35-41.http://www.ncbi.nlm.nih.gov/pubmed/7792813?tool=bestpractice.com
FH-II 的遗传学基础尚不明确。在一个包含 8 名患者的大家庭中,没有发现 FH-II 表型与 CYP11B2、AT1(1 型血管紧张素 II 受体)或 MEN 1(多发性内分泌肿瘤 1 型)基因的多态性存在联系的证据,[41]Torpy DJ, Gordon RD, Lin J-P, et al. Familial hyperaldosteronism type-II: description of a large kindred and exclusion of the aldosterone synthase (CYP11B2) gene. J Clin Endocrinol Metab. 1998;83:3214-3218.http://jcem.endojournals.org/cgi/content/full/83/9/3214http://www.ncbi.nlm.nih.gov/pubmed/9745430?tool=bestpractice.com[42]Torpy DJ, Gordon RD, Stratakis CA. Linkage analysis of familial hyperaldosteronism type II - absence of linkage of the gene encoding the AII receptor type 1. J Clin Endocrinol Metab. 1998;83:1046.http://jcem.endojournals.org/cgi/content/full/83/3/1046http://www.ncbi.nlm.nih.gov/pubmed/9506777?tool=bestpractice.com但全基因组扫描显示与染色体 7p22 上某基因位点相连锁。[43]Lafferty AR, Torpy D, Stowasser M, et al. A novel genetic locus for low-renin hypertension: familial hyperaldosteronism type-II maps to chromosome 7 (7p22). J Med Genet. 2000;37:831-835.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1734468/pdf/v037p00831.pdfhttp://www.ncbi.nlm.nih.gov/pubmed/11073536?tool=bestpractice.com通过对澳大利亚另外 2 个家庭、南美洲的一个家庭和意大利的 2 个家庭的连锁性研究,进一步为 FH-II 涉及 7p22 位点提供了证据。[44]So A, Duffy D, Gordon R, et al. Familial hyperaldosteronism type II is linked to the chromosome 7p22 region but also shows predicted genetic heterogeneity. J Hypertens. 2005;23:1477-1484.http://www.ncbi.nlm.nih.gov/pubmed/16003173?tool=bestpractice.com
最近2008 年首次报告,一个患有PA 和显著束状带增生的家系[11]Geller DS, Zhang J, Wisgerhof MV, et al. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 2008;93:3117-3123.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2515083/http://www.ncbi.nlm.nih.gov/pubmed/18505761?tool=bestpractice.com被确定患有家族性醛固酮增多症 III 型 (FH-III),[12]Mulatero P. A new form of hereditary primary aldosteronism: familial hyperaldosteronism type III. J Clin Endocrinol Metab. 2008;93:2972-2974.http://jcem.endojournals.org/content/93/8/2972.longhttp://www.ncbi.nlm.nih.gov/pubmed/18685118?tool=bestpractice.com其有KCNJ5(编码钾通道)生殖系 突变。[45]Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768-772.http://www.ncbi.nlm.nih.gov/pubmed/21311022?tool=bestpractice.comKCNJ5生殖系突变导致极少见的双侧 PA,并且与发病早有关(年龄 <18 岁),但根据遗传性基因突变的类型其严重程度不同(从轻度至明显的 PA)。[13]Stowasser M. Primary aldosteronism and potassium channel mutations. Curr Opin Endocrinol Diabetes Obes. 2013;20:170-179.http://www.ncbi.nlm.nih.gov/pubmed/23426162?tool=bestpractice.com在 CACNA1H(编码电压门控钙通道)内发生的生殖细胞系突变也见于 10 岁前发作原发性醛固酮增多症的少数个体中。[46]Scholl UI, Stölting G, Nelson-Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. 2015;4:e06315.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4408447/http://www.ncbi.nlm.nih.gov/pubmed/25907736?tool=bestpractice.com在 22 个大且明显的散发醛固酮生成腺瘤中,还确定了8 个KCNJ5 体细胞突变。[45]Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768-772.http://www.ncbi.nlm.nih.gov/pubmed/21311022?tool=bestpractice.com据其他团队报告,KCNJ5 体细胞突变存在于 30% 至 40% 的醛固酮生成腺瘤中。[47]Boulkroun S, Beuschlein F, Rossi GP, et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension. 2012;59:592-598.http://www.ncbi.nlm.nih.gov/pubmed/22275527?tool=bestpractice.com[48]Azizan EA, Murthy M, Stowasser M, et al. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension. 2012;59:587-591.http://www.ncbi.nlm.nih.gov/pubmed/22252394?tool=bestpractice.comAPA 中,发现了ATP1A1(编码 Na+/K+ ATP 酶 α-亚基)、ATP2B3(Ca2+ ATP 酶钙通道)和 CACNA1D(编码电压依赖性钙离子通道)体细胞突变,但其比例较小(分别为 5%、2% 和 11%)。[49]Beuschlein F, Boulkroun S, Osswald A, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet. 2013;45:440-444.http://www.ncbi.nlm.nih.gov/pubmed/23416519?tool=bestpractice.com[50]Scholl UI, Goh G, Stölting G, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. 2013;45:1050-1054.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3876926/http://www.ncbi.nlm.nih.gov/pubmed/23913001?tool=bestpractice.com[51]Azizan EA, Poulsen H, Tuluc P, et al. Somatic mutations in ATP1A1 and CACNA1D
underlie a common subtype of adrenal hypertension. Nat Genet. 2013;45:1055-1060.http://www.ncbi.nlm.nih.gov/pubmed/23913004?tool=bestpractice.com
 [Figure caption and citation for the preceding image starts]: 醛固酮腺瘤来自 Michael Stowasser 博士的个人收集;经获准使用 [Citation ends].根据其与血管紧张素无反应(如在传统Conn肿瘤中的表现)还是有反应,可对醛固酮生成腺瘤进行进一步分类。其中,反应是指血浆醛固酮在过夜卧床后直立 2 到 3 小时期间或者在输注血管紧张素 II期间相对于基线至少升高 50%。
[Figure caption and citation for the preceding image starts]: 醛固酮腺瘤来自 Michael Stowasser 博士的个人收集;经获准使用 [Citation ends].根据其与血管紧张素无反应(如在传统Conn肿瘤中的表现)还是有反应,可对醛固酮生成腺瘤进行进一步分类。其中,反应是指血浆醛固酮在过夜卧床后直立 2 到 3 小时期间或者在输注血管紧张素 II期间相对于基线至少升高 50%。