病因学
Wilms 瘤是具有遗传异质性的肿瘤。[34]Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461-470.http://www.ncbi.nlm.nih.gov/pubmed/18311776?tool=bestpractice.com可能为遗传性或偶发性。遗传研究已发现可在泌尿生殖道正常发育中起重要作用的候选肿瘤抑制基因,[39]Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268-273.http://www.ncbi.nlm.nih.gov/pubmed/9781906?tool=bestpractice.com例如 11p13 基因座上的 WT1 基因,11p15 基因座上的 WT2 基因。这些基因的失活可能是 Wilms 瘤发展中的早期遗传事件。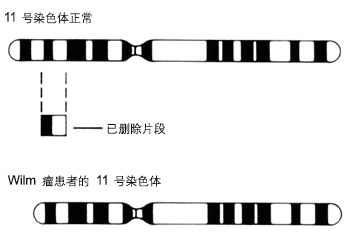 [Figure caption and citation for the preceding image starts]: 11 号染色体缺失国家癌症研究所 (National Cancer Institute);经许可后使用 [Citation ends].1p 和 16q 处的杂合性丢失 (LOH) 被视为最终导致 Wilms 瘤的继发性事件。[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[33]Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270-1273.http://jnci.oxfordjournals.org/cgi/content/full/99/16/1270http://www.ncbi.nlm.nih.gov/pubmed/17686827?tool=bestpractice.com[34]Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461-470.http://www.ncbi.nlm.nih.gov/pubmed/18311776?tool=bestpractice.com[40]Wittmann S, Zirn B, Alkassar M, et al. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer. 2007;46:163-170.http://www.ncbi.nlm.nih.gov/pubmed/17099873?tool=bestpractice.com[41]Zirn B, Samans B, Wittmann S, et al. Target genes of the WNT/beta-catenin pathway in Wilms tumors. Genes Chromosomes Cancer. 2006;45:565-574.http://www.ncbi.nlm.nih.gov/pubmed/16575872?tool=bestpractice.com[42]Zirn B, Hartmann O, Samans B, et al. Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int J Cancer. 2006;118:1954-1962.http://www.ncbi.nlm.nih.gov/pubmed/16287080?tool=bestpractice.com[43]Dome JS, Bockhold CA, Li SM, et al. High telomerase RNA expression level is an adverse prognostic factor for favorable-histology Wilms' tumor. J Clin Oncol. 2005;23:9138-9145.http://www.ncbi.nlm.nih.gov/pubmed/16172460?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: 11 号染色体缺失国家癌症研究所 (National Cancer Institute);经许可后使用 [Citation ends].1p 和 16q 处的杂合性丢失 (LOH) 被视为最终导致 Wilms 瘤的继发性事件。[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[33]Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270-1273.http://jnci.oxfordjournals.org/cgi/content/full/99/16/1270http://www.ncbi.nlm.nih.gov/pubmed/17686827?tool=bestpractice.com[34]Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461-470.http://www.ncbi.nlm.nih.gov/pubmed/18311776?tool=bestpractice.com[40]Wittmann S, Zirn B, Alkassar M, et al. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer. 2007;46:163-170.http://www.ncbi.nlm.nih.gov/pubmed/17099873?tool=bestpractice.com[41]Zirn B, Samans B, Wittmann S, et al. Target genes of the WNT/beta-catenin pathway in Wilms tumors. Genes Chromosomes Cancer. 2006;45:565-574.http://www.ncbi.nlm.nih.gov/pubmed/16575872?tool=bestpractice.com[42]Zirn B, Hartmann O, Samans B, et al. Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int J Cancer. 2006;118:1954-1962.http://www.ncbi.nlm.nih.gov/pubmed/16287080?tool=bestpractice.com[43]Dome JS, Bockhold CA, Li SM, et al. High telomerase RNA expression level is an adverse prognostic factor for favorable-histology Wilms' tumor. J Clin Oncol. 2005;23:9138-9145.http://www.ncbi.nlm.nih.gov/pubmed/16172460?tool=bestpractice.com
在某些先天性过度生长综合征(例如 Beckwith-Wiedemann 综合征、孤立的偏身肥大、帕尔曼综合征、巨脑畸形综合征以及 Simpson-Golabi-Behmel 综合症)中,发展 Wilms 瘤的风险增加。[5]Green DM, Breslow NE, Beckwith JB, et al. Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol. 1993;21:188-192.http://www.ncbi.nlm.nih.gov/pubmed/8095320?tool=bestpractice.com先天性非过度生长综合征,包括孤立的无虹膜症、18 三体、WAGR 综合症(即 Wilms 瘤、无虹膜症、泌尿生殖器异常、智力障碍)、布卢姆综合征以及 Denys-Drash 综合征也与其发展相关。[6]Heppe RK, Koyle MA, Beckwith JB. Nephrogenic rests in Wilms tumor patients with the Drash syndrome. J Urol. 1991;145:1225-1228.http://www.ncbi.nlm.nih.gov/pubmed/1851891?tool=bestpractice.com[7]Breslow NE, Norris R, Norkool PA, et al. Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2003;21:4579-4585.http://www.ncbi.nlm.nih.gov/pubmed/14673045?tool=bestpractice.com有先天性泌尿生殖器异常(例如尿道下裂、外阴性别不明或隐睾症)的儿童也有发展 Wilms 瘤的倾向。[8]Mahmood A, Ghafoor T, Badsha S. Wilms' tumour: presentation and treatment. J Coll Physicians Surg Pak. 2004;14:142-145.http://www.ncbi.nlm.nih.gov/pubmed/15228845?tool=bestpractice.com[9]Breslow NE, Olson J, Moksness J, et al. Familial Wilms' tumor: a descriptive study. Med Pediatr Oncol. 1996;27:398-403.http://www.ncbi.nlm.nih.gov/pubmed/8827065?tool=bestpractice.com[10]van den Heuvel-Eibrink MM, Grundy P, Graf N, et al. Characteristics and survival of 750 children diagnosed with a renal tumor in the first seven months of life: a collaborative study by the SIOP/GPOH/SFOP, NWTSG, and UKCCSG Wilms tumor study groups. Pediatr Blood Cancer. 2008;50:1130-1134.http://www.ncbi.nlm.nih.gov/pubmed/18095319?tool=bestpractice.com尽管如此,只有一小部分病例 (10%) 与先天畸形综合征相关或有阳性家族史。[3]Breslow N, Olshan A, Beckwith JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21:172-181.http://www.ncbi.nlm.nih.gov/pubmed/7680412?tool=bestpractice.com[6]Heppe RK, Koyle MA, Beckwith JB. Nephrogenic rests in Wilms tumor patients with the Drash syndrome. J Urol. 1991;145:1225-1228.http://www.ncbi.nlm.nih.gov/pubmed/1851891?tool=bestpractice.com[44]Nishi M, Miyake H, Takeda T, et al. Congenital malformations and childhood cancer. Med Pediatr Oncol. 2000;34:250-254.http://www.ncbi.nlm.nih.gov/pubmed/10742060?tool=bestpractice.com
除遗传因素之外,许多流行病学研究已尝试发现可能与 Wilms 瘤直接相关的特异性出生前环境因素,包括父亲的职业(例如焊接或机械)或父亲对农药、电离辐射、咖啡因或染发产品的接触。[3]Breslow N, Olshan A, Beckwith JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21:172-181.http://www.ncbi.nlm.nih.gov/pubmed/7680412?tool=bestpractice.com[45]Cooney MA, Daniels JL, Ross JA, et al. Household pesticides and the risk of Wilms tumor. Environ Health Perspect. 2007;115:134-137.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=17366833http://www.ncbi.nlm.nih.gov/pubmed/17366833?tool=bestpractice.com[46]Saddlemire S, Olshan AF, Daniels JL, et al. Breast-feeding and Wilms tumor: a report from the Children's Oncology Group. Cancer Causes Control. 2006;17:687-693.http://www.ncbi.nlm.nih.gov/pubmed/16633916?tool=bestpractice.com[47]Neville H, Ritchey ML, Shamberger RC, et al. The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg. 2002;37:1134-1137.http://www.ncbi.nlm.nih.gov/pubmed/12149688?tool=bestpractice.com[48]Green DM, Cotton CA, Malogolowkin M, et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine and actinomycin D: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer. 2007;48:493-499.http://www.ncbi.nlm.nih.gov/pubmed/16547940?tool=bestpractice.com[49]Campbell AD, Cohn SL, Reynolds M, et al. Treatment of relapsed Wilms' tumor with high-dose therapy and autologous hematopoietic stem-cell rescue: the experience at Children's Memorial Hospital. J Clin Oncol. 2004;22:2885-2890.http://www.ncbi.nlm.nih.gov/pubmed/15254057?tool=bestpractice.com[50]Kim TH, Zaatari GS, Baum ES, et al. Recurrence of Wilms tumor after apparent cure. J Pediatr. 1985;107:44-49.http://www.ncbi.nlm.nih.gov/pubmed/2989472?tool=bestpractice.com但这些研究大部分尚无明确结论。[4]Bunin GR, Meadows AT. Epidemiology and Wilms tumor: approaches and methods. Med Pediatr Oncol. 1993;21:169-171.http://www.ncbi.nlm.nih.gov/pubmed/8383277?tool=bestpractice.com[22]Ward EM, Thun MJ, Hannan LM, et al. Interpreting cancer trends. Ann N Y Acad Sci. 2006;1076:29-53.http://www.ncbi.nlm.nih.gov/pubmed/17119192?tool=bestpractice.com[51]Crump C, Sundquist J, Sieh W, et al. Perinatal risk factors for Wilms tumor in a Swedish national cohort. Eur J Epidemiol. 2014;29:191-197.http://www.ncbi.nlm.nih.gov/pubmed/24510487?tool=bestpractice.com
病理生理学
Wilms 瘤的形成被认为是一个经过癌前病变的多步骤过程。肾源性残留是正常肾组织小叶内或小叶周边原始肾胚基的微观灶,源于肾干细胞。它们见于 30%-40% 单侧和所有双侧 Wilms 瘤的正常肾组织,[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[39]Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268-273.http://www.ncbi.nlm.nih.gov/pubmed/9781906?tool=bestpractice.com[52]Coppes MJ, Beckwith JB. Clinical approach to renal lesions in children with multiple nephrogenic rests. Med Pediatr Oncol. 2000;35:73-74.http://www.ncbi.nlm.nih.gov/pubmed/10881011?tool=bestpractice.com[53]Park S, Bernard A, Bove KE, et al. Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms' tumour. Nat Genet. 1993;5:363-367.http://www.ncbi.nlm.nih.gov/pubmed/8298644?tool=bestpractice.com以及先天性过度生长或非过度生长综合征儿童。[6]Heppe RK, Koyle MA, Beckwith JB. Nephrogenic rests in Wilms tumor patients with the Drash syndrome. J Urol. 1991;145:1225-1228.http://www.ncbi.nlm.nih.gov/pubmed/1851891?tool=bestpractice.com[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[54]Beckwith JB. Management of incidentally encountered nephrogenic rests. J Pediatr Hematol Oncol. 2007;29:353-354.http://www.ncbi.nlm.nih.gov/pubmed/17551393?tool=bestpractice.com[55]Perlman E, Dijoud F, Boccon-Gibod L. Nephrogenic rests and nephroblastomatosis (in French). Ann Pathol. 2004;24:510-515.http://www.ncbi.nlm.nih.gov/pubmed/15785399?tool=bestpractice.com这支持肾源性残留是前期病变这一观点。
第一遗传模型或“二次突变假说”(相继出现的 2 次限速突变事件)假定发展遗传性 Wilms 瘤的儿童出生时在基因的一个等位基因中有组成性合子形成前 DNA 突变(这是初始事件),且继该事件之后发生另一个相同或不同基因座的遗传事件,导致肿瘤形成。[56]Knudson AG Jr, Strong LC. Mutation and cancer: a model for Wilms' tumor of the kidney. J Natl Cancer Inst. 1972;48:313-324.http://www.ncbi.nlm.nih.gov/pubmed/4347033?tool=bestpractice.com[57]Knudson AG Jr. The genetic predisposition to cancer. Birth Defects Orig Artic Ser. 1989;25:15-27.http://www.ncbi.nlm.nih.gov/pubmed/2655739?tool=bestpractice.com[58]Knudson AG Jr, Strong LC. Letter: familial Wilm's tumor. Am J Hum Genet. 1975;27:809-810.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=173187http://www.ncbi.nlm.nih.gov/pubmed/173187?tool=bestpractice.com后续研究已识别与遗传性和偶发性 Wilms 瘤都相关的特定染色体区域的多个推定肿瘤抑制基因或致癌基因。这些染色体区域(及基因)包括:11p13(WT1 和 PAX6 基因)和 11p15(WT2 基因、IGFII、H19、p57kip2)以及 17q (FWT1)、19q (FWT2)、16q、1p、5q14、22q12、Xp22、2p24、rs807624、11q14 和 17p (p53)。[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[33]Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270-1273.http://jnci.oxfordjournals.org/cgi/content/full/99/16/1270http://www.ncbi.nlm.nih.gov/pubmed/17686827?tool=bestpractice.com[34]Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461-470.http://www.ncbi.nlm.nih.gov/pubmed/18311776?tool=bestpractice.com[40]Wittmann S, Zirn B, Alkassar M, et al. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer. 2007;46:163-170.http://www.ncbi.nlm.nih.gov/pubmed/17099873?tool=bestpractice.com[41]Zirn B, Samans B, Wittmann S, et al. Target genes of the WNT/beta-catenin pathway in Wilms tumors. Genes Chromosomes Cancer. 2006;45:565-574.http://www.ncbi.nlm.nih.gov/pubmed/16575872?tool=bestpractice.com[43]Dome JS, Bockhold CA, Li SM, et al. High telomerase RNA expression level is an adverse prognostic factor for favorable-histology Wilms' tumor. J Clin Oncol. 2005;23:9138-9145.http://www.ncbi.nlm.nih.gov/pubmed/16172460?tool=bestpractice.com[59]Osterheld MC, Caron L, Meagher-Villemure K. Role of DNA content analysis and immunohistochemistry in the evaluation of the risk of unfavourable outcome in Wilms' tumours. Anticancer Res. 2008;28:751-756.http://www.ncbi.nlm.nih.gov/pubmed/18507016?tool=bestpractice.com[60]Ogawa O, Eccles MR, Szeto J, et al. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms' tumour. Nature. 1993;362:749-751.http://www.ncbi.nlm.nih.gov/pubmed/8097018?tool=bestpractice.com[61]Reeve AE, Eccles MR, Wilkins RJ, et al. Expression of insulin-like growth factor-II transcripts in Wilms' tumour. Nature. 1985;317:258-260.http://www.ncbi.nlm.nih.gov/pubmed/2995817?tool=bestpractice.com[62]Koufos A, Hansen MF, Copeland NG, et al. Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature. 1985;316:330-334.http://www.ncbi.nlm.nih.gov/pubmed/2991766?tool=bestpractice.com[63]Grundy P, Koufos A, Morgan K, et al. Familial predisposition to Wilms' tumour does not map to the short arm of chromosome 11. Nature. 1988;336:374-376.http://www.ncbi.nlm.nih.gov/pubmed/2848199?tool=bestpractice.com[64]Francke U. Molecular genetics. A gene for Wilms tumour? Nature. 1990;343:692-694.http://www.ncbi.nlm.nih.gov/pubmed/2154698?tool=bestpractice.com[65]Turnbull C, Perdeaux ER, Pernet D, et al. A genome-wide association study identifies susceptibility loci for Wilms tumor. Nat Genet. 2012;44:681-684.http://www.ncbi.nlm.nih.gov/pubmed/22544364?tool=bestpractice.com研究表明母体 IGF-II 基因印记缺失导致的 IGF-II 致癌基因异常表达可能是 Wilms 瘤最常见的表观遗传改变。[33]Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270-1273.http://jnci.oxfordjournals.org/cgi/content/full/99/16/1270http://www.ncbi.nlm.nih.gov/pubmed/17686827?tool=bestpractice.com此外,小叶内肾源性残留更可能与在源于相同宿主的 Wilms 瘤中发现的相同基因突变相关。[32]Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer. 2006;47:260-267.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16700047http://www.ncbi.nlm.nih.gov/pubmed/16700047?tool=bestpractice.com[39]Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268-273.http://www.ncbi.nlm.nih.gov/pubmed/9781906?tool=bestpractice.com[66]Malogolowkin M, Cotton CA, Green DM, et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer. 2008;50:236-241.http://www.ncbi.nlm.nih.gov/pubmed/17539021?tool=bestpractice.com[67]Feusner JH, Ritchey ML, Norkool PA, et al. Renal failure does not preclude cure in children receiving chemotherapy for Wilms tumor: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer. 2008;50:242-245.http://www.ncbi.nlm.nih.gov/pubmed/17458877?tool=bestpractice.com[68]Felgenhauer JL, Barce JM, Benson RL, et al. No excess of early onset cancer in family members of Wilms tumor patients. Cancer. 2001;92:1606-1612.http://www.ncbi.nlm.nih.gov/pubmed/11745239?tool=bestpractice.com[69]Shamberger RC, Guthrie KA, Ritchey ML, et al. Surgery-related factors and local recurrence of Wilms tumor in National Wilms Tumor Study 4. Ann Surg. 1999;229:292-297.http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=10024113http://www.ncbi.nlm.nih.gov/pubmed/10024113?tool=bestpractice.com[70]Hennigar RA, O'Shea PA, Grattan-Smith JD. Clinicopathologic features of nephrogenic rests and nephroblastomatosis. Adv Anat Pathol. 2001;8:276-289.http://www.ncbi.nlm.nih.gov/pubmed/11556536?tool=bestpractice.com
总之,这些研究表明,肾源性残留和肿瘤都可能源于普通胚胎肾干细胞的克隆,且导致肿瘤形成的这些病变中可能发生多个连续遗传和表观遗传事件,例如肿瘤抑制基因的失活及 1p 和 16q 处的 LOH。[53]Park S, Bernard A, Bove KE, et al. Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms' tumour. Nat Genet. 1993;5:363-367.http://www.ncbi.nlm.nih.gov/pubmed/8298644?tool=bestpractice.com
经证明,在染色体区 1q21.1-q31.3 获取的复制数 (CN) 与复发以及染色体臂 1p、1q、3p、3q 和 14q 处的等位基因失衡显著相关。[71]Perotti D, Spreafico F, Torri F, et al. Genomic profiling by whole-genome single nucleotide polymorphism arrays in Wilms tumor and association with relapse. Genes Chromosomes Cancer. 2012;51:644-653.http://www.ncbi.nlm.nih.gov/pubmed/22407497?tool=bestpractice.com[72]Gratias EJ, Jennings LJ, Anderson JR, et al. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer. 2013;119:3887-3894.http://www.ncbi.nlm.nih.gov/pubmed/23983061?tool=bestpractice.com
使用此内容应接受我们的免责声明。
